铜族金属与完整及氮掺杂石墨烯的相互作用
基于广义梯度密度泛函理论和周期平板模型, 研究了铜族金属单原子和双原子簇与完整及氮掺杂石墨烯的结合情况. 结果表明, 氮掺杂后石墨烯的电子结构特性由半金属性变为金属性; 铜族金属在完整及石墨型氮掺杂石墨烯上的吸附较弱, 结合能约为 0.5 eV, 而在吡啶型氮掺杂和吡咯型氮掺杂石墨烯上有较强的化学吸附, 结合能一般大于 1 eV; 吡咯型氮掺杂后的构型不稳定, 金属原子及簇与包含该结构的石墨烯衬底作用时会使其向吡啶型氮掺杂转变, 并最终得到基于吡啶型氮掺杂的稳定吸附构型. Mulliken 电荷布居分析显示, 吸附在吡啶型氮掺杂石墨烯上的金属单原子与金属双原子簇带电性质相反. 态密度及轨道分析表明, Cu 与吡啶型氮掺杂石墨烯空位处留有悬挂键的三个原子成键, 而 Au 与其中两个 C 原子成键.
完整石墨烯具有理想的二维晶体结构, 由碳原子以 sp2 杂化形式连接成一平面单原子层, 理论厚度为 0.35 nm. 石墨烯独特的结构使其表现出许多奇异的物理化学性质; 其极高的比表面积可以减少催化剂活性组分的团聚, 增加有效活性位, 从而降低催化剂的成本. 因此, 以石墨烯为载体的催化剂体系被广泛研究; 理论研究显示, Cu 及 Au嵌入石墨烯能有效催化氧化 CO.采用直接裁剪、控制层数、化学功能化、异种原子化学掺杂等方法改变石墨烯的结构可以调控其电子特性. 氮原子掺杂是一种有效改变石墨烯电子结构的方法. 氮原子与碳原子尺寸相当, 且有五个价电子, 可以和碳原子形成强的共价键, 目前在实验室已经实现了氮掺杂石墨烯的可控制备.氮掺杂石墨烯的电子结构发生了变化, 用作催化剂
载体时性能优于完整石墨烯. 需要指出的是, 虽然完整石墨烯为载体的催化剂的有关报道较多, 但氮掺杂石墨烯负载催化剂的研究却不多. 多相贵金属催化剂广泛应用于石油化工、精细化工、环保催化、生命及生物化学等领域. 研究表明, 当贵金属颗粒以纳米级形态存在于催化剂中时, 真空技术网石墨烯栏目(http://www.chvacuum.com/graphene/)认为往往表现出非常高的催化活性和选择性.
本文使用密度泛函理论, 研究了铜族金属单原子和双原子簇与完整及氮掺杂石墨烯的相互作用.首先比较完整石墨烯和三种氮掺杂石墨烯的电子结构特征, 考察氮掺杂后石墨烯电子结构的变化; 然后, 计算 Cu 族金属单原子及双原子簇在各种衬底不同位置进行吸附时的结合能, 得出最稳定的构型、最大结合能及相应的吸附高度; 最后, 分析相互作用过程中成键及电子转移情况.
1、模型和计算方法
计算模型选用单层六方石墨烯 4 × 4 超晶胞结构, 超晶胞内包含有 32 个原子, 如图 1 所示. (a) 为完整的石墨烯, 用 PG 表示; (b) 为石墨型 N 原子掺杂, 即 N 原子取代完整石墨烯的一个 C 原子, 此种结构用 NG1 表示; (c) 为吡啶型 N 原子掺杂, 即移去完整石墨烯晶胞的两个 C 原子, 然后由一个 N 原子占据其中一个 C 原子的位置, 形成一个含 N 的六元环, 该结构用 NG2 表示; (d) 为吡咯型 N 原子掺杂, 先移除两个 C 原子, 然后由一个 N 原子与空位附近的四个 C 原子形成五元环, 该结构用 NG3 表示. NG2 和 NG3 的超晶胞包含 31 个原子. 三种 N掺杂后的石墨烯仍然保持平面结构, 这均已在实验上得到证实. 显然, 由于移去了 C 原子, NG2和 NG3 分别各有两个 C 原子只成了两个键, 留有悬挂键. 这两个特殊 C 原子与同样留有悬挂键的掺杂N 原子形成的三角形中心区域称为空位, 该位置为吸附时的高活性位.
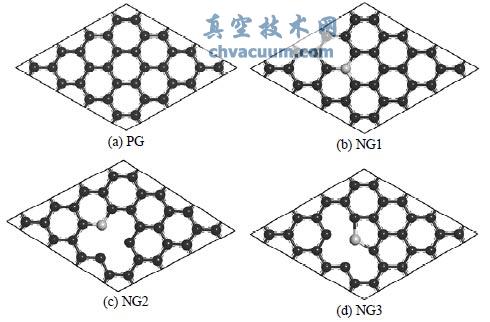
图 1 四种衬底的 4 × 4 超晶胞模型
所有计算均采用非限制性 Kohn-Sham 方法, 由Dmol3 软件包实现[33,34]. 使用广义梯度近似 (GGA),泛函选用 PW91; 考虑到重金属元素的相对论效应,Cu, Ag, Au 原子内层电子由半核赝势 (DSPP) 来处理, 价电子波函数采用双数值基组加极化函数展开(DNP), C 和 N 原子采用全电子基组; 结构优化时自洽迭代过程布里渊区积分 K 点选取为 3 × 3 × 1 格点; 计算能带和态密度时 K 点设置为 15 × 15 × 1;相邻两层石墨烯之间的距离设为 1.2 nm, 确保层间相互作用足够小. 采用以上参数优化的石墨烯中C–C 键长为 0.1423 nm, 与文献[35,36]值 (0.1424 和0.1425 nm) 较为一致 . 在本文中, 结合能定义为吸附前后各物质总能量能量之差.
2、结论
采用密度泛函理论研究了 Cu 族金属单原子和双原子簇与完整及氮掺杂石墨烯的结合情况. 结果表明, 氮掺杂后石墨烯的电子结构由半金属性向金属性转变. 通过分析稳定构型、结合能、吸附高度和电荷布居情况, 得到 Cu 族金属单原子和双原子簇与完整及氮掺杂石墨烯相互作用的规律: 二者与PG 和 NG1 的结合能力均较差, 是物理吸附或者弱的化学吸附; 在四种衬底上的结合能力几乎均为Cu > Au > Ag, 且金属混合双原子簇在衬底上结合总是结合能力强的金属原子与衬底成键时为最稳定构型; Cu 总是能与衬底空位处留有悬挂键的三原子成键, Ag 只有在单原子吸附时才能与空位处三原子成键, Au 在任何情况下都只能与留有悬挂键的两个 C 原子成键. 吡咯型氮掺杂不稳定, 金属原子及簇与包含该结构的石墨烯衬底发生作用时会使其向吡啶型氮掺杂转变, 并最终得到基于吡啶型氮掺杂的稳定吸附构型; 二者与 NG2 作用后电子转移方向不同, 其中由金属单原子向 NG2 的电子转移可能对某些催化反应的研究有重要参考意义.