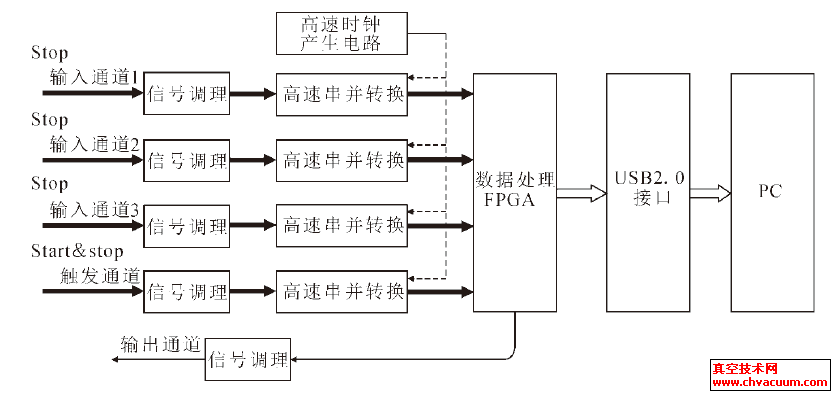
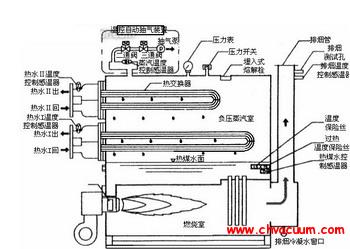
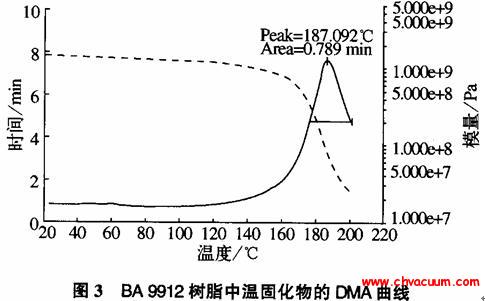
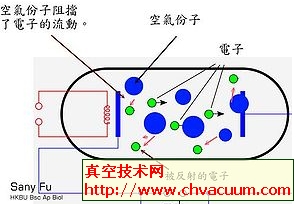
Ce掺杂SnO2材料结构和光学性质的研究
基于密度泛函理论的第一性原理平面波超软赝势方法,用广义梯度近似(GGA)PBE交换相关泛函,研究稀土Ce掺杂的SnO2材料能带结构和光学性质,对纯SnO2和掺杂后的能带结构、态密度以及光学性质进行了分析对比。结果表明,掺杂后材料均属直接跃迁半导体,Ce的4f轨道进入导带使导带底向低能端移动,禁带宽度变小,吸收光谱与介电函数虚部谱线相对应,吸收谱发生红移,峰强减弱,各峰值与电子跃迁吸收有关。因此,本文可对接下来的实验研究有一定借鉴作用。
SnO2是一种具有金红石结构Ⅳ-Ⅵ族宽禁带半导体材料,空间群为P42/mnm,常温下禁带宽度为3.6eV,激子束缚能为130meV,在多个领域如气敏元件、太阳能电池、电极材料及半导体元件等方面有着广泛的应用前景,是一种重要的无机功能材料。SnO2因其内部结构疏松致密度低等特点在高敏感材料中应用不是很理想,对SnO2掺杂改性研究成为主要研究方向,如Cu/CuO掺杂制备高性能气敏材料、掺杂Ag制备薄膜传感器材料。此外,掺杂元素对SnO2电子结构、磁学性能以及晶体结构和形貌方面的影响也有诸多研究。杜娟等采用密度泛函平面波赝势法对含Ⅲ族杂质SnO2超晶胞的电子结构进行了研究,Barbarat等运用第一性原理研究了SnO2的光电等性质,发现了化学键与光学性质的关联性,徐剑等对F掺杂SnO2电子结构、能带结构和态密度进行模拟计算,分析了掺杂对晶体性质和光学吸收边的影响。稀土元素具有独特的4f电子结构,由于有外层5s和5p轨道的屏蔽,温度和晶体场对4f电子跃迁几乎没有影响,因此已被广泛运用于电磁器件和发光材料的生产中,是SnO2掺杂体系研究的新热点。Gu等人发现Ce掺杂SnO2后增强了紫色发光峰,随Ce浓度增大发光峰强度先增加后减弱。由于有关稀土掺杂SnO2材料的性质和机理尚不完全清楚,因此从理论上进行深入研究,对于SnO2的应用将具有重要的意义。本文采用第一性原理的方法研究了Ce掺杂SnO2材料,并对其电子结构和光学性质进行了计算,对其性能进行了分析,为以后相关研究提供相应的参考。
1、构建模型及计算方法
基于密度泛函理论的第一性原理,采用周期性边界条件,用广义梯度近似(GGA)下PBE来处理电子间的交换关联能,SnO2为金红石结构,空间群为P42/mnm,对称性D4h-l4,SnO2属体心四方晶系,其原胞包含两个Sn原子和四个O原子,Sn原子占据四方体顶点和体心位置。对Ce掺杂SnO2构建2×2×2的超晶胞结构进行模拟计算,取SnO2晶格常数实验值α=β=γ=90°,a=b=0.4737nm,c=0.3186nm,如图1所示。平面波截断能设为350eV,自洽迭代过程中使用的k点数为3×3×4。计算过程迭代进行,自洽收敛精度为1×10-5eV/atom。计算含有Ce掺杂的SnO2时,对超晶胞均进行了结构优化,作用在各个原子上的力均小于0.05eV/nm,计算中考虑的价电子组态有:O2s22p4;Sn5s25p2;Ce4f15d15p26s2,计算都在倒易空间中进行。
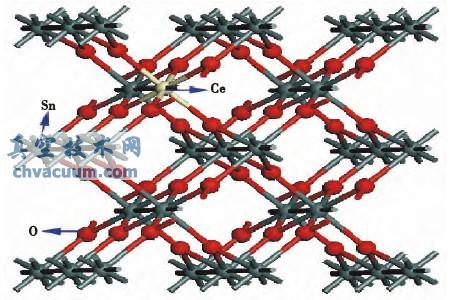
图1 Ce掺杂SnO2超晶胞结构
2、结果与分析
2.1、能带结构与态密度分析
由图2(a)可看出导带底和价带顶位于Brillouin区G点处,说明SnO2是直接带隙半导体,带隙为1.64eV,虽然采用GGA近似,但计算的带隙远低于实验值3.6eV,这是由于采用DFT计算晶体结构时,禁带普遍偏低。对SnO2晶体而言,主要是计算中过高估计Sn5s的能量,造成Sn5s与O2p相互作用的增大,使价带带宽增大,带隙偏低,但这不影响对SnO2电子结构的理论分析。Ce外层电子轨道为4f15d16s2,含有1个f电子和1个d电子,可由图2(b)看出,在价带顶和导带底之间没有引入中间能级,与纯SnO2相比,掺Ce后导带的能带变得更加密集,带间起伏变得平缓,并且整个价带和导带都向低能端移动。这说明Ce原子的掺入导致不同原子之间的相互作用变强,使禁带宽度变窄,这就说明电子从价带激发到导带所需的能量变小,因而导致吸收带的红移。
3、结论
运用第一性原理研究了稀土元素Ce掺杂SnO2的能带结构与光学性质,讨论了Ce对SnO2体系的能带结构和光学性质的影响。掺杂后材料属直接跃迁半导体,Ce的4f轨道进入导带使导带底向低能端移动,禁带宽度变小,吸收光谱与介电函数虚部谱线相对应,吸收谱发生红移,峰强减弱,各峰值与电子跃迁吸收有关。在0eV~9eV范围内,由于Ce掺杂使SnO2能带结构发生变化,形成了不同类型的跃迁,使吸收率高于纯SnO2,即在紫外光区的透过率降低。